



The CRISPRCasFinder program enables the easy detection of CRISPRs and cas genes in user-submitted sequence data (allows sequences up to 50 Mo otherwise download standalone program). This is an update of the CRISPRFinder program with improved specificity and indication on the CRISPR orientation. MacSyFinder is used to identify cas genes, the CRISPR-Cas type and subtype.
A maximal repeat is a repeat with no possible extension to the right or the left without incurring a mismatch.
Maximal repeats have interesting computational properties since they can be computed in linear time using a suffix-tree-based algorithm and their number is linear (at most equal to the sequence length). A CRISPR structure is a succession of maximal repeats (the direct repeats) separated by the spacers. CRISPRFinder uses this property to find possible localizations of CRISPRs. Finding the maximal repeats is done with VMatch which is the upgrade of REPuter (Kurtz 1999 [1]) based on an efficient implementation of enhanced suffix arrays (Abouelhoda 2004 [2]).
The main idea of the CRISPRFinder program is to find possible CRISPR localizations and then to check if these regions contain a cluster that meets CRISPR structure standards.
The following figure illustrates various conformations of repeats within CRISPR arrays:
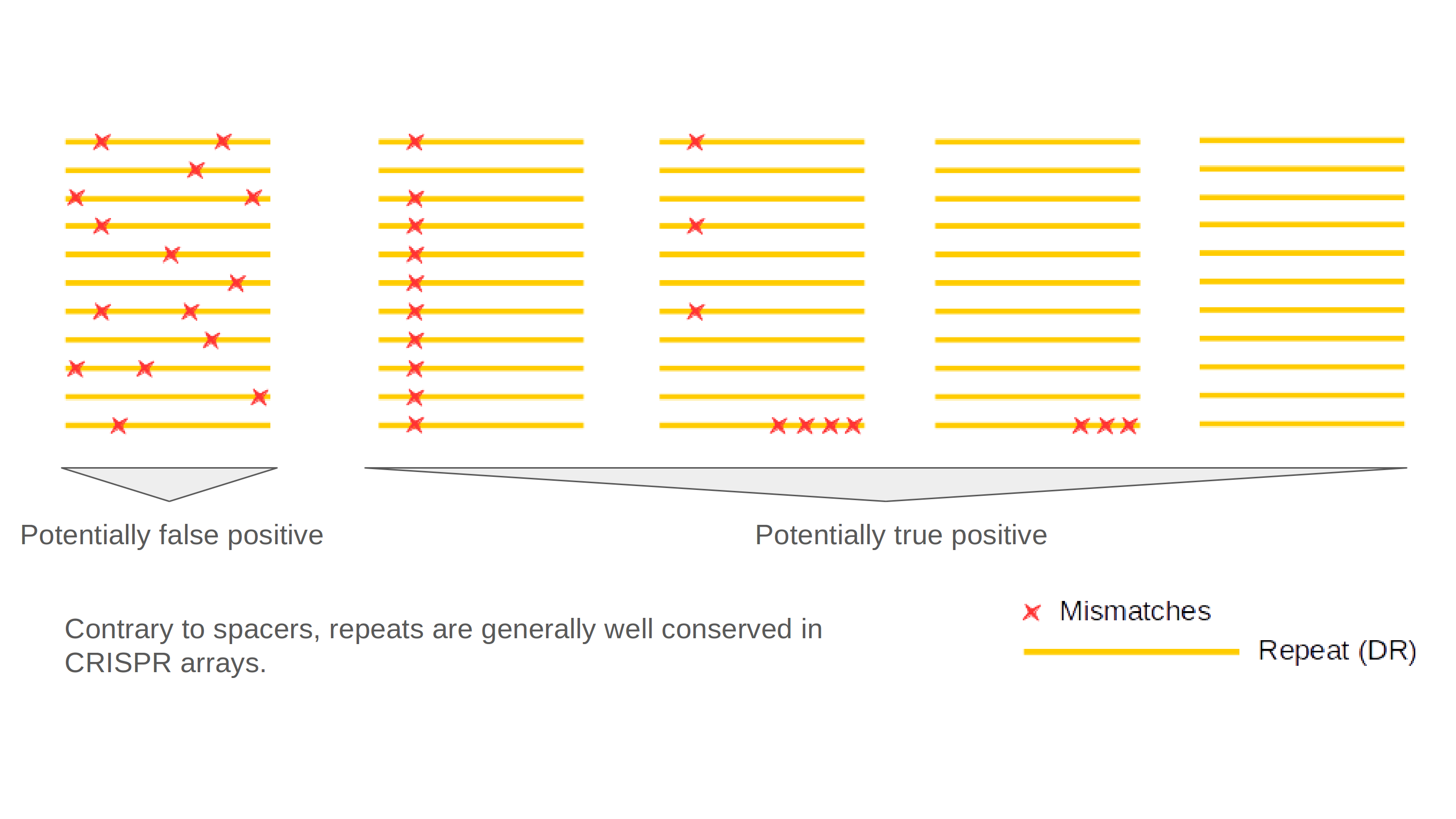
To identify the potential orientation of CRISPRs, two tests have been implemented. The CRISPRDirection program predicts orientation by comparison to a curated dataset (updated in May 2017) of consensus repeats. The orientation is shown in the summary table as a + (if the region is the left flanking) or - sign (if the region is the right flanking) ND (Not determined) is shown when no prediction can be made. In addition, the AT% is calculated in 100bp flanking the array on both sides. The region with the higher AT% is considered as a leader and the result is shown in the detailed results.
The first step consists in the identification of open reading frames (ORF) with Prodigal [4]. Then these ORFs are analysed by the MacSyFinder program by HMM search of a library of known Cas proteins [5]. The Cas type and subtype are found by analysis of clusters of Cas.
The search for CAS will return no result if the cluster of genes is not complete and therefore not functional. In such cases some cas genes may be present and will be detected by using the General clustering model.
1- Stefan Kurtz, Chris Schleiermacher. REPuter: Fast Computation of Maximal Repeats in Complete Genomes, Bioinformatics 15(5): 426-427, 1999.
2- M.I. Abouelhoda, S. Kurtz, and E. Ohlebusch. Replacing Suffix Trees with Enhanced Suffix Arrays, Journal of Discrete Algorithms, 2:53-86, 2004.
3- Biswas, A., Fineran, P.C. and Brown, C.M. Accurate computational prediction of the transcribed strand of CRISPR non-coding RNAs, Bioinformatics 30(13): 1805-1813, 2014.
4- Hyatt,D. Prodigal: prokaryotic gene recognition and translation initiation site identification, BMC Bioinformatics, 11: 119, 2010
5- Abby, S. S.,Neron, B., Menager, H., Touchon, M., Rocha, E. P. : MacSyFinder: a program to mine genomes for molecular systems with an application to CRISPR-Cas systems, PloSOne, 9: e110726, 2014.